����1984��ͨ����
��Hatch-Waxman��
����
����������ҩ����ANDA(AbbreviatedNew Drug Application)
����Ҫ�ݽ��ٴ�ǰ���ٴ�������֤����ȫ�Ժ���Ч��
���������ѧ��֤������ҩ��ԭ��ҩ�������Ч��
��
NCE����ҩ����
��NewDrug application��
���NDA��
��������4���ĵ�һ��
������ҩ���̿��ύ����PIV������ANDA����
����1�����벢�ɹ������ҩ�ʸ��
���ɻ��180����г���ռ����
������һ���ύ����ķ���ҩ�����Թ�����180��Ķ�ռ��
��������һ����ǰ�ύ��ANDA���붼���ᱻ����
������սר����PI��PII��PIII������Ҫ���һ��
����NCE��5�����ݶ�ռ�������ܹ��ύ
��
��
����Ҫ�ݽ���Щ�ļ�
�����߿��Ը���FDA�����������ļ�����ANDA������
��
��ANDA Submissions �C Contentand Format of
Abbreviated New Drug Applications��
��ANDA FILLING CHECKLI��
FDA������õ��ӵݽ��ķ�ʽ
����ΪOfficeof Generic Drugs(OGD)û���㹻�Ŀռ������ֽ�ʵ��걨�ļ�
���ο�FDA������ָ��
��ProvidingRegulatory Submissions in Electronic
Format ��Certain Human PharmaceuticalProduct Applications and Related
Submissions Using the eCTD Specifications��
��ANDA����Ҳ����ȡeCTD�ĸ�ʽ����������
��
Module 1
��Ҫ����Administrative����Ϣ,��ر�����Դ�FDA��վ������http://www.fda.gov/AboutFDA/ReportsManualsForms/Forms/default.htm��
��Ҫ˵��һ��ר��������
����������Ҫ����I.ANDA�ķ��ƶ���û��ר����Ϣ
��II��
ר���ѹ���
��III��
ר������ij�չ���
��IV��
ר����Ч
�����߱���ANDA���ַ���ר��
��дһ��ר��֤��
��������������Ҫ��սר��
������IV������
������ҩ��������Ҫ���ύANDA��20����֪ͨԭ��ҩ������
��ԭ��ҩ�����߿�����45������Ժ�ύ����
��FDA�Ը�ANDA�������Զ��Ӻ�30����
��
Module 2 CTD Summaries, ��Ҫ����2.3 QOS�Լ�2.7 Clinical summary
(Bioequivalence); ����ҩ�����������Ŀ�ѧ�Ժͷ����Ե�Ҫ��
��FDA������ʵʩ��һ��QbR��Questionbased
Review��
������,��������Ҫ����FDA�ṩ��QbR��ģ����дQoS��
Module 3 CMC����
��
Module 4һ��ANDA����Ҫ�ύ����������
��
Module 5 ������������
���ⲿ���ύ����Ϣ�ǻ��FDA���Ĺؼ�����
��
һ�����ڽ��������бȽϷ���ҩ��orangebook�й涨��RLD(�α��Ƽ�)���������ö�����������
���о����ܰ�����ʳǰ��ҩƷ���������ö�����
��
��
��������
ANDA����CDER�����ķ���ҩ�칫��
��Officeof Generic Drugs��
������
������ҩ�����յ�ҩƷ��������orangebook�е�RLD��
����ҩ������˾��Ҫ���걨������֤������Ƶ�ҩƷ��RLD������ͬ�Ļ��Գɷ�
��ͬ��������������
����ͬ�ļ���
�����
����ҩ;��
���÷�������
����ǩ˵�������Ҫ�����������ͬ
���Լ�֤������ҩ��RLD���������Ч��
��
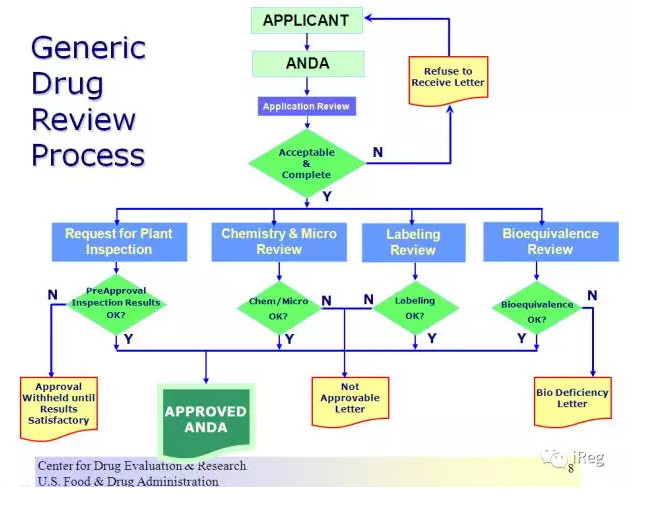
��ͼ�Ƿ���ҩ��������:
ANDA�����ĸ��ؼ�������
�������������
��ҩƷ��˵��������
����ѧ/��������
���Լ��������ֳ����
������
�������Ч�������ǹؼ�����
��
��
����ʱ��
ANDA����һ���ύ
��FDAһ������180���ڽ�������
��������approved��approvable����notapprovable��֪ͨ
��